We May Not Understand Control Groups
active placebo, clinical trials, control group effects, placebo effect, side effects
It’s well known that randomized trials are some of the most efficient ways to make causal inferences and to determine how much something (an intervention) differs from the comparator (some sort of control). Random assignment helps make these goals easier by minimizing selection bias and making the distribution of prognostic factors between groups random (not balanced).[@Zhao2018-yf]
Discussions (similar to the one above) praising the efficiency of randomized trials are widespread, however, few of these discussions take a close look at some of the common assumptions that individuals hold regarding randomized trials. And unfortunately, these common assumptions may be based on outdated evidence and simplistic ideas.
The Placebo Effect Isn’t What We Often Think
For example, in placebo-controlled trials, many individuals make the assumption that much of the improvement seen in the control group over time is due to the placebo effect, as modeled by the graph below.

However, inquiries into this topic have yielded contradictory results, in that the placebo effect may not be as powerful as we assume.
One systematic review[@Hrobjartsson2001-if] that looked at 130 clinical trials concluded the following,
“We found little evidence in general that placebos had powerful clinical effects. Although placebos had no significant effects on objective or binary outcomes, they had possible small benefits in studies with continuous subjective outcomes and for the treatment of pain. Outside the setting of clinical trials, there is no justification for the use of placebos.”
Thus, the placebo effect had some small effects in areas where it is difficult to objectively measure a phenomenon and where there is a higher likelihood for encountering measurement error. However, measurement error is not the only thing that could explain some of the improvements seen in control groups, which we often attribute to the placebo effect.
We simply need to reread two sections of a paper by the very anesthesiologist who popularized the placebo effect[@Beecher1955-yc] and who claimed that it had the ability to put patients’ conditions into remission.
Here, Henry Beecher (the anesthesiologist) claims that placebo effects are powerful:
“It is evident that placebos have a high degree of therapeutic effectiveness in treating subjective responses, decided improvement, interpreted under the unknowns technique as a real therapeutic effect, being produced in 35.2 ± 2.2% of cases….
Placebos have not only remarkable therapeutic power but also toxic effects. These are both subjective and objective. The reaction (psychological) component of suffering has power to produce gross physical change.”
Then he reports another observation from his patients.
“The evidence is that placebos are most effective when the stress is greatest.”
If you measured someone’s stress (let’s say objectively, via cortisol measurements) and they were extremely stressed and if you followed up after a certain period of time, it would be very likely that the next measurement of their stress (cortisol levels) would be less than the first measurement.
A slightly reverse situation would likely apply too: if you measured someone’s happiness (in some theoretical objective way) and they were extremely happy, it’s very likely that their next measurement of happiness would not be so high. This phenomenon is known as regression to the mean,[@Senn2011-fj] and it can explain why many individuals who are in an abnormal state, feel slightly more normal over time.
We can see this displayed below in the graph of simulated blood pressure data[@Atkinson2015-dd] where the change in blood pressure is predicted by baseline blood pressure. The individuals with higher blood pressure at baseline experienced a larger decrease in blood pressure over time.

This could possibly explain why Beecher observed that placebo was most effective for situations in which stress was the greatest.
It may not have been just the patients’ minds that led to remission of symptoms, rather the combination of regressing towards an average from a more extreme value, the placebo effect, along with the subjectivity and measurement error of the instruments may have led to this phenomenon of experiencing relief as a function of time.[@mcdonaldHowMuchPlacebo1983]
This isn’t to say that placebo effects are nonexistent or that they’re not relevant, they still are because they serve as controls for various other phenomena that come along with them. For example, it seems very unlikely that in a clinical trial that recruits healthy participants and aims to improve their intelligence with an experimental drug, that regression to the mean could explain the improvements seen in the control group.
In a trial aiming to improve biomarkers in healthy participants, could regression towards the mean or noise explain all the variance we see in the control group? Probably not. It seems very feasible that the placebo effect can result in changes to both subjective and objective outcomes, irrespective of the effects of regression to the mean and measurement error, however, I’m inclined to believe that many of the claims regarding the placebo effect may be exaggerated.
Thus, a more accurate way to think of control group effects would be to incorporate phenomena such as regression to the mean, the placebo effect, and other factors, as shown below.

Of course, the proportion of these individual effects would likely vary depending on the scenario(s). Here, the proportions of effects in the control group reflect my beliefs about what contributes to improvements seen in sick individuals who are followed over time.
While the model above is certainly an improvement over the first bar graph in the article all the way above, it too may also be simplistic… and may even be wrong.[@Kube2017-jt] I’ll first discuss some theoretical examples and then present some evidence.
The Additive Model Is Too Simple
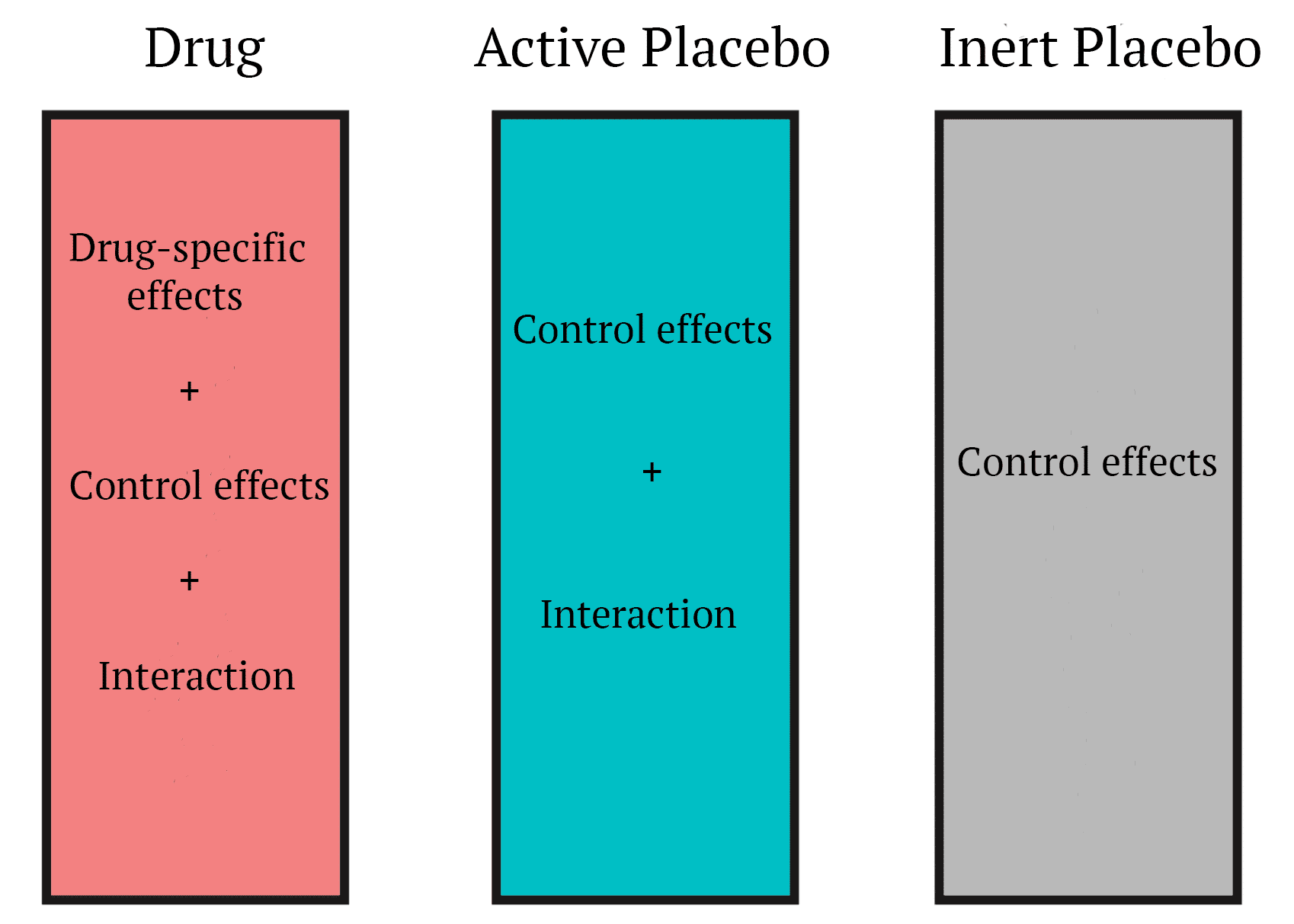
If we add up all the possible effects that lead to improvements in the control group, such as regression to the mean, the placebo effect, measurement error, unspecific effects etc, we can call this giant block (the blue block) the “total control group effect”, at least for the sake of this discussion. This is shown in the bar graph below. Notice that it has a total effect of 6.

Again, here we are assuming that all the possible things that lead to improvements in the control group are represented by the blue bar. Thus, we design a clinical trial with the assumption that the total control group effect, AKA that represented by the blue bar, will also be the same in the treatment group. This is the “Additive Model” because it simply adds the treatment effect (red bar) on top of the control effect (blue bar), giving a total effect of 10. We subtract the total effect of the treatment group (10) from the total control group effect (6) to get:
Treatment-specific effect (4) = Total treatment group effect (10) - Total control group effect (6)
Therefore, we estimate that the drug has an effect of 4 whereas all the effects that contribute to the control result in an effect of 6. In the form of a linear model, we would model this as y = constant + b(group), where b is the difference between the treatment group and the control group, and group would be coded as 0 or 1, etc.
This idea may be too simplistic as it assumes that the control effect (the blue bar) will be constant between groups.
However, several studies have shown disparities in placebo/control effects between groups, with placebo/control effects sometimes being larger in control groups than in treatment groups.[@Finnerup2010-kw; @Finnerup2015-ga; @Rutherford2014-ye]
Experimental studies have also found that the sum of the drug effect and the placebo effect is often larger than the total treatment effect.[@Hammami2010-ud; @Lund2014-if] This doesn’t seem to support the additive model, where we assume the total control effect is constant in both groups.
It doesn’t take into account that there may be an interaction[@Corraini2017-xi; @Rothman1974-ky] that occurs between the control group effects and the treatment effects, especially if there are possible factors that could lead the participants in the clinical trial to discover that they’ve been allocated to the treatment group.
Thus, a more inclusive model of these possible factors, in the form of a linear model, would be:
y = *constant + βgroup + βinteractio**n(s) + ϵ*

These interactions could result in many scenarios, but let’s look at two simple ones.
If a participant in a drug trial is aware of the side effects of the drug beforehand, and receives the drug and experiences side effects, he/she may now have some confirmation that they’ve received the actual intervention, and may overestimate their improvement in a subjective outcome and/or may experience placebo/physiological mechanisms that overlap and interact, thus amplifying the control effects. The overall treatment group effect would increase. This would be a synergistic interaction[@Rothman1974-ky], as shown below. Notice the control group has a total effect of 6, while the drug group has a total effect of 13.

Now let’s say in another scenario, a patient in a drug trial was unaware of the possible side effects of the drug and experienced some side effects, which has somehow convinced the participant that they may not improve, or the physiological interaction in the mechanisms between the drug and placebo result in negative outcomes. This may lead to a reduction in the placebo effect and a reduction in other unspecific control effects. This would be an antagonistic interaction[@Rothman1974-ky], as shown below. It’s reduced the overall treatment group effect from 13 to 8.

Thus, when we design clinical trials with comparators, we’re often assuming that the treatment-specific effect can be estimated by taking the difference between the total treatment group effect and the total control group effect, because we assume the control effect is constant between groups. But again, as shown above, this may be far too simplistic.
Possible Directions
So what’s the solution? How can we design clinical trials to better account for these possible interactions between the intervention and the control group effects? A potential solution that’s been put forth by several researchers over the years is the balanced-placebo design.[@Atlas2012-lr; @Hammami2010-ud; @Kube2017-jt; @Lund2014-if; @Schenk2014-pa] The balanced-placebo design is essentially a 2 x 2 factorial design where two groups will receive the drug and two groups will receive placebo.

Out of the two groups receiving the drug, one will be told that they’re receiving placebo, while the other is told they’re receiving the drug. The same would apply to the two groups receiving the placebo. One will be told that they’re receiving the placebo, while the other is told they’re receiving the drug. While this design may account for interactions between control group effects and the intervention, I can’t say I’m much of a fan of it as it requires more groups (therefore more participants) and doesn’t use blinding. It may also be unethical.
A very simple solution to me (out of several proposed designs) is the use of an “active placebo.”[@Kube2017-jt] Active placebos are substances that don’t yield any beneficial effects, but are designed to mimic some of the side effects of the intervention (pretty sure a more appropriate term for them would be nocebo). Thus, controlling for the possible interaction(s) that could occur if the participant is affected by the side effects of the intervention. This will make it harder for the participant to know whether they got the intervention or the placebo.
Psychiatrist Scott Alexander of Slate Star Codex and LessWrong is skeptical of active placebos and believes that the research doesn’t support their usage.[@Quitkin2000-st] In his article about selective serotonin reuptake inhibitors he writes,
“In other words, active placebo research has fallen out of favor in the modern world. Most studies that used active placebo are very old studies that were not very well conducted. Those studies failed to find an active-placebo-vs.-drug difference because they weren’t good enough to do this. But they also failed to find an active-placebo-vs.-inactive-placebo difference. So they provide no support for the idea that active placebos are stronger than inactive placebos in depression and in fact somewhat weigh against it.”
Scott uses a narrative review from 2000[@Quitkin2000-st] that looked at previous active-placebo trials to support this perspective, but several other studies that have used active placebos have been carried out since
- Several of these are of much higher quality than the studies discussed in the review he cites. A more recent methodological review,[@Jensen2017-jp] published by one of the authors who systematically searched the literature to find that placebo effects were underwhelming in trials with objective outcomes,[@Hrobjartsson2001-if] concludes the following,
Pharmacological active placebo control interventions are rarely used in randomized clinical trials, but they constitute a methodological tool which merits serious consideration. We suggest that active placebos are used more often in trials of drugs with noticeable side effects, especially in situations where the expected therapeutic effects are modest and the risk of bias due to unblinding is high.
Luckily, active placebo research doesn’t seem to have fallen out of favor. Is active placebo research the solution to the problems mentioned above? I’m not sure. There are several other proposed designs that attempt to account for possible interactions that can occur and they may be more practical or less biased. It seems like recognizing that such interactions could occur in the first place would be the first step towards finding a solution.
Acknowledgements: I’d like to thank Andrew Vigotsky for bringing my attention to several of the pieces cited in this article and for offering comments on an early version of this piece.
References
Citation
@online{panda2018,
author = {Panda, Sir and Rafi, Zad},
title = {We {May} {Not} {Understand} {Control} {Groups}},
date = {2018-10-28},
url = {https://lesslikely.com/posts/statistics/control-group-effects.html},
langid = {en}
}